HCN1 epilepsy: From Genetics and Mechanisms to Precision Therapies
August 11, 2023
Abstract found on PubMed
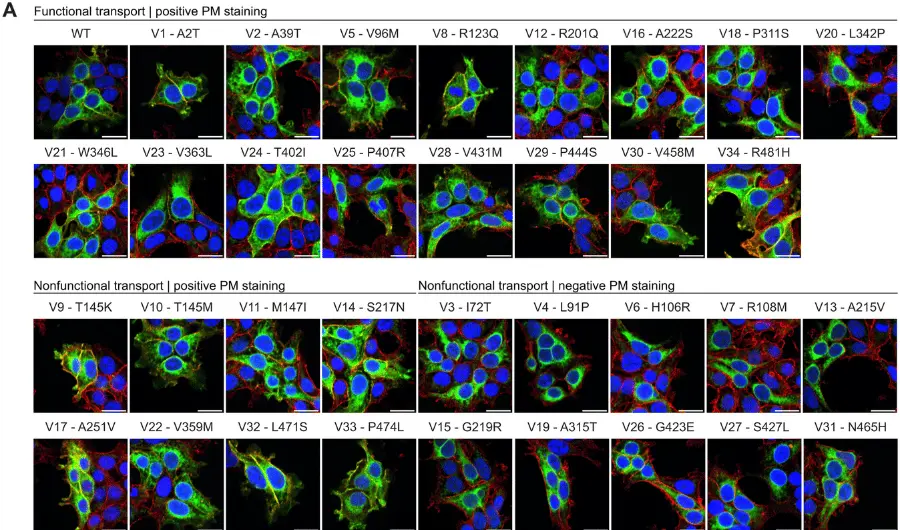
Pathogenic variation in HCN1 is now an established cause of epilepsy and intellectual disability. Variation in HCN1 causes a spectrum of disease with a genotype-phenotype relationship emerging. De novo pathogenic variants that occur in the transmembrane domains of the channel typically cause a cation ‘leak’ that associates with severe developmental and epileptic encephalopathy (DEE). Genotype-phenotype associations for variants that fall outside of the transmembrane domains are less well established but do include milder forms of epilepsy that can be either de novo or inherited. HCN1 DEE mouse models have been generated which recapitulate the seizures and learning difficulties seen in human patients. These mice have also acted as powerful preclinical models which share pharmacoresponsiveness with human HCN1 DEE patients. Data from these mouse models support the conclusion that anti-seizure medications with sodium channel block as their primary mechanism of action should be used with caution in HCN1 DEE. Other comorbidities of HCN1 DEE including retinal dysfunction have also been modelled in HCN1 DEE mice, suggesting HCN1 variants can cause a dramatically reduced sensitivity to light with limited ability to process temporal information. Our understanding of the genetics and pathophysiological mechanisms underlying HCN1 epilepsy has progressed significantly and is already influencing therapy. However, more research effort is needed to fully understand the natural histories of HCN1 epilepsies and to develop precision therapeutic approaches.
More Epilepsy Research News