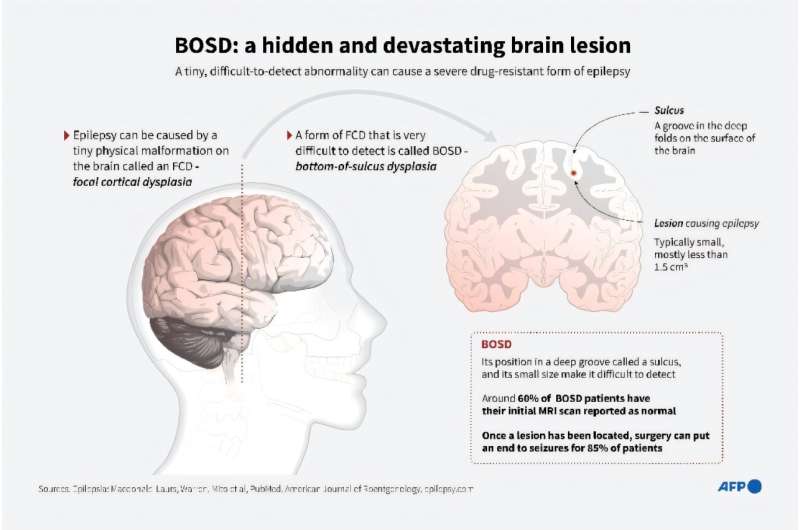
The Relation of Etiology Based on the 2017 ILAE Classification to the Effectiveness of the Ketogenic Diet in Drug-Resistant Epilepsy in Childhood
August 30, 2021
Abstract, originally published in Epilepsia
Objective: To investigate the effectiveness and safety of the ketogenic diet (KD) in drug-resistant epilepsy in childhood in relation to the new 2017 International League Against Epilepsy (ILAE) classification of etiology.
Methods: A consecutive cohort of patients treated with the KD were categorized according to the ILAE classification into known (structural, genetic, metabolic, infectious, and immune-mediated) and unknown etiology. Primary outcome was the frequency of patients achieving seizure freedom with the KD at 3 months, secondary outcomes were seizure reduction >50% at 3 months, and both seizure freedom and seizure reduction >50% at 6, 12 months, and at last follow-up (LFU), and adverse effects. Outcomes were compared between etiology groups.
Results: Etiology was known in 70% (129/183). Outcomes did not differ at 3 months (known vs unknown: seizure freedom 28% vs 33%, seizure reduction 62 vs 67%), but seizure freedom was significantly less frequent in known etiology at 6 months (26% vs 43%) and beyond (22% vs 37%). Logistic regression identified duration of epilepsy, number of previous antiseizure medications (ASMs), and age-appropriate psychomotor development as positive determinants of outcome. Among individual etiology groups, the effectiveness of KD was relatively best for genetic (33% at LFU) and poorest for metabolic etiology (8% at LFU). The small number of patients with infectious and immune-mediated etiology requires larger numbers in each etiology group to corroborate our results. No differences in type and frequency of adverse effects (in 71%) between etiology groups were observed, requiring medical intervention in 21%.
Significance: The ketogenic diet was most effective in genetic and unknown etiology, many unknowns probably represent yet unidentified genetic causes. We recommend consequent diagnostic and genetic work-up to identify etiologies that respond best to the ketogenic diet. The ketogenic diet should be offered early to infants with genetic epilepsy before deterioration of epileptic symptoms and of psychomotor development.
Latest from the Community