A Two-Hit Story: Genetic Mutations Increase the Risk of a ‘Second Hit’ to Developing Severe Epilepsy
February 1, 2019
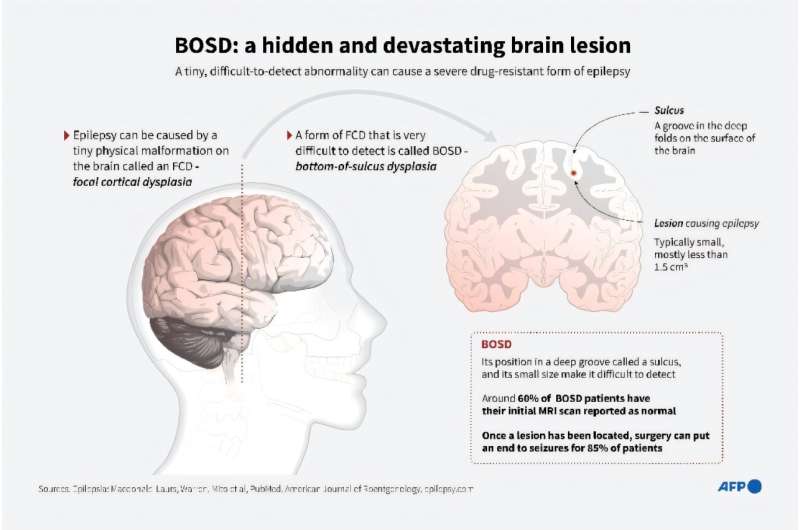
SCN1A (NaV1.1 sodium channel) mutations cause Dravet syndrome (DS) and GEFS+ (which is in general milder), and are risk factors in other epilepsies. Phenotypic variability limits precision medicine in epilepsy, and it is important to identify factors that set phenotype severity and their mechanisms. It is not yet clear whether SCN1A mutations are necessary for the development of severe phenotypes or just for promoting seizures. A relevant example is the pleiotropic R1648H mutation that can cause either mild GEFS+ or severe DS.
Researchers used a R1648H knock-in mouse model (Scn1aRH/+) with mild/asymptomatic phenotype to dissociate the effects of seizures and of the mutation per se. The induction of short repeated seizures, at the age of disease onset for Scn1a mouse models (P21), had no effect in WT mice, but transformed the mild/asymptomatic phenotype of Scn1aRH/+ mice into a severe DS-like phenotype, including frequent spontaneous seizures and cognitive/behavioral deficits. In these mice, we found no major modifications in cytoarchitecture or neuronal death, but increased excitability of hippocampal granule cells, consistent with a pathological remodeling.
Therefore, this study claims to demonstrate for their model that an SCN1A mutation is a prerequisite for a long term deleterious effect of seizures on the brain, indicating a clear interaction between seizures and the mutation for the development of a severe phenotype generated by pathological remodeling. Applied to humans, this result suggests that genetic alterations, even if mild per se, may increase the risk of second hits to develop severe phenotypes.
Latest from the Community